Protein-ligand Docking tutorial using BioExcel Building Blocks (biobb)
– Fpocket Version –
This tutorial aims to illustrate the process of protein-ligand docking, step by step, using the BioExcel Building Blocks library (biobb). The particular example used is the Mitogen-activated protein kinase 14 (p38-α) protein (PDB code 3HEC, https://doi.org/10.2210/pdb3HEC/pdb), a well-known Protein Kinase enzyme, in complex with the FDA-approved Imatinib, (PDB Ligand code STI, DrugBank Ligand Code DB00619), a small molecule kinase inhibitor used to treat certain types of cancer.
The tutorial will guide you through the process of identifying the active site cavity (pocket) without previous knowledge, and the final prediction of the protein-ligand complex.
Please note that docking algorithms, and in particular, AutoDock Vina program used in this tutorial, are non-deterministic. That means that results obtained when running the workflow could be diferent from the ones we obtained during the writing of this tutorial (see AutoDock Vina manual). We invite you to try the docking process several times to verify this behaviour.
Settings
Biobb modules used
biobb_io: Tools to fetch biomolecular data from public databases.
biobb_structure_utils: Tools to modify or extract information from a PDB structure file.
biobb_chemistry: Tools to perform chemoinformatics processes.
biobb_vs: Tools to perform virtual screening studies.
Auxiliary libraries used
Conda Installation
git clone https://github.com/bioexcel/biobb_wf_virtual-screening.git
cd biobb_wf_virtual-screening
conda env create -f conda_env/environment.yml
conda activate biobb_wf_virtual-screening
jupyter-notebook biobb_wf_virtual-screening/notebooks/fpocket/biobb_wf_virtual-screening_fpocket.ipynb
Pipeline steps
![]()
Initializing colab
The two cells below are used only in case this notebook is executed via Google Colab. Take into account that, for running conda on Google Colab, the condacolab library must be installed. As explained here, the installation requires a kernel restart, so when running this notebook in Google Colab, don’t run all cells until this installation is properly finished and the kernel has restarted.
# Only executed when using google colab
import sys
if 'google.colab' in sys.modules:
import subprocess
from pathlib import Path
try:
subprocess.run(["conda", "-V"], check=True)
except FileNotFoundError:
subprocess.run([sys.executable, "-m", "pip", "install", "condacolab"], check=True)
import condacolab
condacolab.install()
# Clone repository
repo_URL = "https://github.com/bioexcel/biobb_wf_virtual-screening.git"
repo_name = Path(repo_URL).name.split('.')[0]
if not Path(repo_name).exists():
subprocess.run(["mamba", "install", "-y", "git"], check=True)
subprocess.run(["git", "clone", repo_URL], check=True)
print("⏬ Repository properly cloned.")
# Install environment
print("⏳ Creating environment...")
env_file_path = f"{repo_name}/conda_env/environment.yml"
subprocess.run(["mamba", "env", "update", "-n", "base", "-f", env_file_path], check=True)
print("🎨 Install NGLView dependencies...")
subprocess.run(["mamba", "install", "-y", "-c", "conda-forge", "nglview==3.0.8", "ipywidgets=7.7.2"], check=True)
print("👍 Conda environment successfully created and updated.")
# Enable widgets for colab
if 'google.colab' in sys.modules:
from google.colab import output
output.enable_custom_widget_manager()
# Change working dir
import os
os.chdir("biobb_wf_virtual-screening/biobb_wf_virtual-screening/notebooks/fpocket")
print(f"📂 New working directory: {os.getcwd()}")
Input parameters
Input parameters needed:
pdb_code: PDB code of the experimental complex structure (if exists).
In this particular example, the p38α structure in complex with the Imatinib drug was experimentally solved and deposited in the PDB database under the 3HEC PDB code, https://doi.org/10.2210/pdb3HEC/pdb. The protein structure from this PDB file will be used as a target protein for the docking process, after stripping the small molecule. An APO structure, or any other structure from the p38α cluster 100 (sharing a 100% of sequence similarity with the p38α structure) could also be used as a target protein. This structure of the protein-ligand complex will be also used in the last step of the tutorial to check how close the resulting docking pose is from the known experimental structure.
ligandCode: Ligand PDB code (3-letter code) for the small molecule (e.g. STI, DrugBank Ligand Code DB00619).
In this particular example, the small molecule chosen for the tutorial is the FDA-approved drug Imatinib (PDB Code STI, DrugBank Ligand Code DB00619), a type of cancer growth blocker, used in diferent types of leukemia.
pockets_dir: Name of a folder to write temporary files.
import nglview
import ipywidgets
pdb_code = "3HEC" # P38 + Imatinib
ligand_code = "STI" # Imatinib
pockets_dir = "pockets"
Fetching PDB structure
Downloading PDB structure with the protein molecule from the PDBe database.
Alternatively, a PDB file can be used as starting structure.
Building Blocks used:
Pdb from biobb_io.api.pdb
from biobb_io.api.pdb import pdb
download_pdb = "download.pdb"
prop = {
"pdb_code": pdb_code,
"filter": ["ATOM", "HETATM"]
}
pdb(output_pdb_path=download_pdb,
properties=prop)
Visualizing 3D structure
Visualizing the downloaded/given PDB structure using NGL.
Note (and try to identify) the Imatinib small molecule (STI) and the detergent (β-octyl glucoside) (BOG) used in the experimental reservoir solution to obtain the crystal.
view = nglview.show_structure_file(download_pdb, default=True)
view.center()
view._remote_call('setSize', target='Widget', args=['','600px'])
view
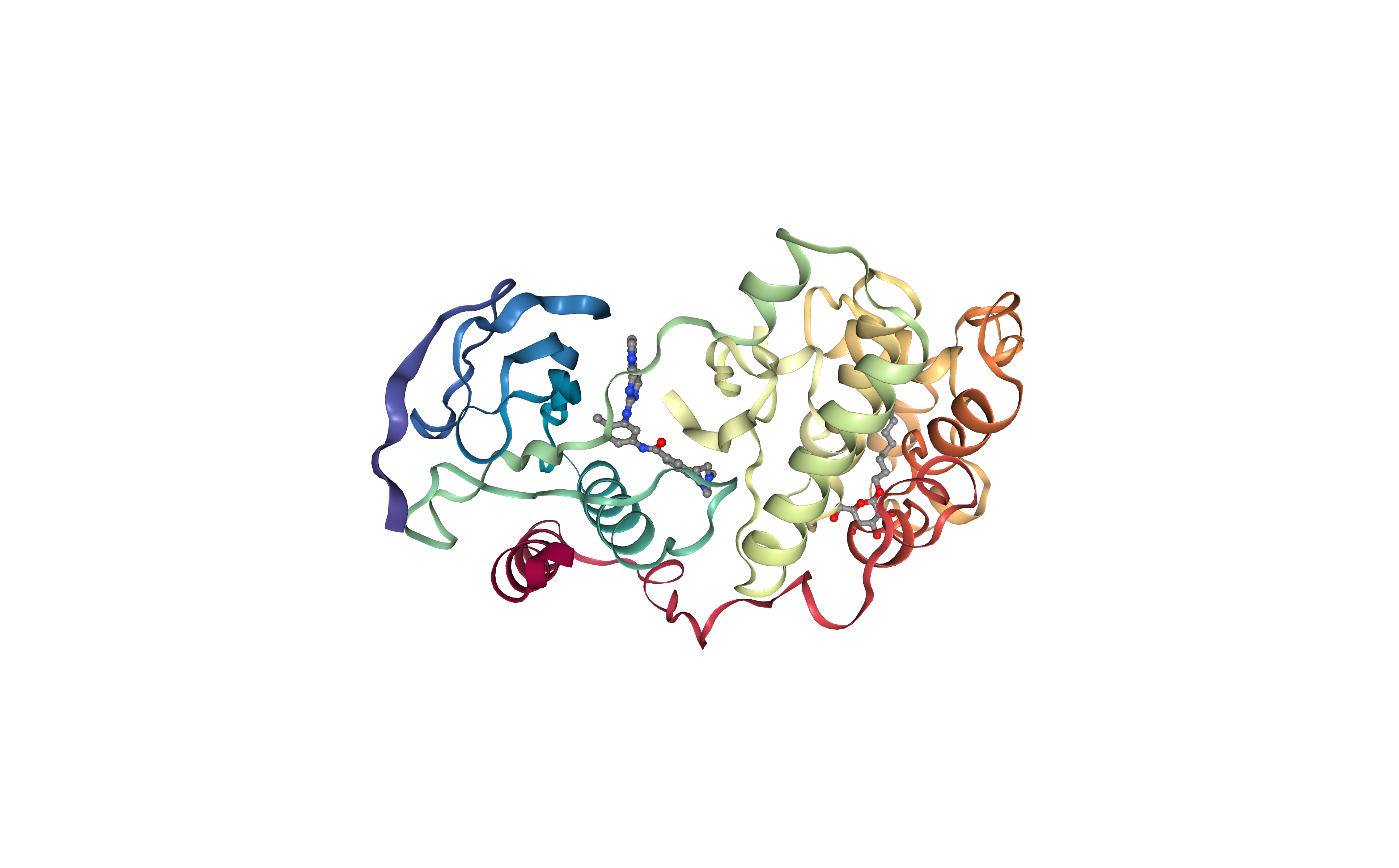
Extract Protein Structure
Extract protein structure from the downloaded PDB file. Removing any extra molecule (ligands, ions, water molecules).
The protein structure will be used as a target in the protein-ligand docking process.
Building Blocks used:
extract_molecule from biobb_structure_utils.utils.extract_molecule
from biobb_structure_utils.utils.extract_molecule import extract_molecule
pdb_protein = "pdb_protein.pdb"
extract_molecule(input_structure_path=download_pdb,
output_molecule_path = pdb_protein)
Visualizing 3D structure
Visualizing the downloaded/given PDB structure using NGL.
Note that the small molecules included in the original structure are now gone. The new structure only contains the protein molecule, which will be used as a target for the protein-ligand docking.
view = nglview.show_structure_file(pdb_protein, default=False)
view.add_representation(repr_type='cartoon',
selection='not het',
colorScheme = 'atomindex')
view.center()
view._remote_call('setSize', target='Widget', args=['','600px'])
view
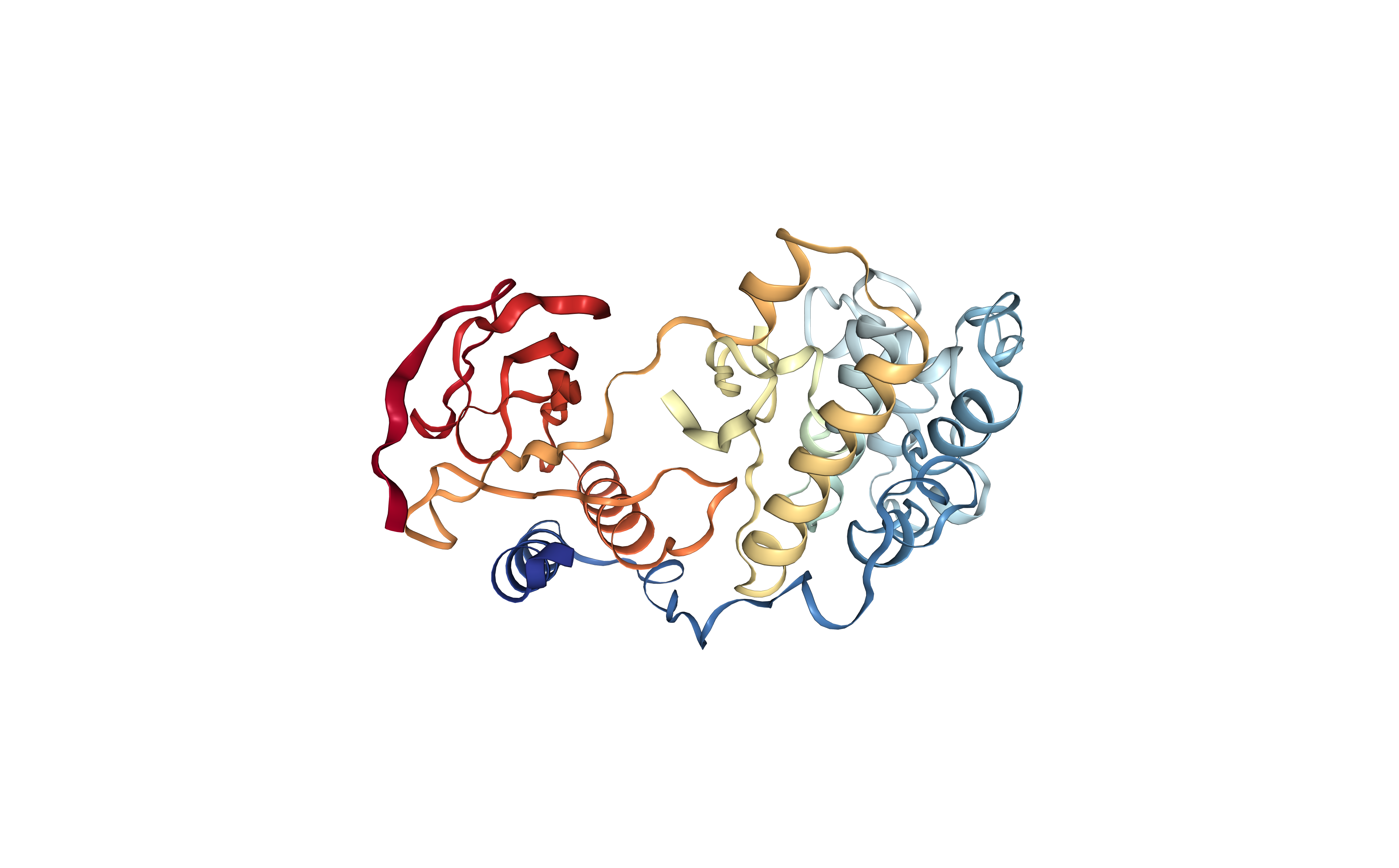
Computing Protein Cavities (fpocket)
Computing the protein cavities (pockets) using the well-known fpocket tool.
These cavities will be then used in the docking procedure to try to find the best region of the protein surface where the small molecule can bind.
Although in this particular example we already know the binding site region, as we started from a protein-ligand complex structure where the ligand was located in the same binding site as Imatinib is binding, this is not always the case. In the cases where we do not know these regions, fpocket will help us identifying the possible binding sites of our target protein.
fpocket input parameters, such as minimum and maximum radius (in Angstroms) the alpha spheres might have in a binding pocket can be adjusted (min_radius, max_radius) . Parameters used in this particular example are 3Å for the minimum radius and 6Å for the maximum radius. The minimum number of alpha spheres a pocket must contain in order to figure in the results is also adjusted to 35. See the fpocket manual for more information.
Building Blocks used:
fpocket_run from biobb_vs.fpocket.fpocket_run
from biobb_vs.fpocket.fpocket_run import fpocket_run
fpocket_all_pockets = "fpocket_all_pockets.zip"
fpocket_summary = "fpocket_summary.json"
prop = {
"min_radius": 3,
"max_radius": 6,
"num_spheres": 35
}
fpocket_run(input_pdb_path=pdb_protein,
output_pockets_zip = fpocket_all_pockets,
output_summary=fpocket_summary,
properties=prop)
Checking fpocket output (json)
Checking the fpocket output from the json file. Every pocket has a separated entry in the json output, with information such as: score, druggability score, volume, hydrophobicity, polarity or flexibility.
import json
with open(fpocket_summary, 'r') as json_file:
data = json.load(json_file)
print(json.dumps(data, indent=4))
Filtering Protein Cavities (fpocket output)
Filtering the protein cavities (pockets) identified by fpocket.
In this particular example, the biggest cavities, with a volume between 800 and 2000 ($Å^{3}$), big enough volume to fit the input small molecule, are selected.
Building Blocks used:
fpocket_filter from biobb_vs.fpocket.fpocket_filter
from biobb_vs.fpocket.fpocket_filter import fpocket_filter
fpocket_filter_pockets = "fpocket_filter_pockets.zip"
prop = {
"volume": [800, 2000]
}
fpocket_filter(input_pockets_zip=fpocket_all_pockets,
input_summary = fpocket_summary,
output_filter_pockets_zip=fpocket_filter_pockets,
properties=prop)
Extract selected pockets (cavities)
Extract the selected pockets (cavities) from the filtered list (zip file, fpocket_filter_pockets).
Writing the information in the pockets_dir folder.
Also saving the list of PDB files (protein residues forming the pocket) and PQR files (cavity, pocket), to be used in following visualization step.
import os
import shutil
from pathlib import Path, PurePath
import zipfile
if Path(pockets_dir).exists(): shutil.rmtree(pockets_dir)
os.mkdir(pockets_dir)
with zipfile.ZipFile(fpocket_filter_pockets, 'r') as zip_ref:
zip_ref.extractall(pockets_dir)
path_pockets = [str(i) for i in Path(pockets_dir).iterdir()]
path_pockets_pdb = [str(i) for i in Path(pockets_dir).iterdir() if PurePath(i).suffix == '.pdb']
path_pockets_pqr = [str(i) for i in Path(pockets_dir).iterdir() if PurePath(i).suffix == '.pqr']
Visualizing selected pockets (cavities)
Visualizing the selected pockets (cavities) from the filtered list using NGL viewer.
Protein residues forming the cavity are represented in random-colored surfaces. Pockets are represented in a blue-colored mesh. Different pockets are identified with a floating label.
import re
import random
# random colors for cavities
r = lambda: random.randint(0,255)
# load structure
view = nglview.NGLWidget()
c = view.add_component(nglview.FileStructure(pdb_protein))
# load cavities (d) and pockets (p) and create pocketNames list
c = {}
p = {}
pocketNames = []
for pock in path_pockets:
g = re.findall('(?:pocket)(\d+)(?:_\w+)\.(\w+)', pock)
i = g[0][0]
suff = g[0][1]
if not [item for item in pocketNames if ('pocket' + i) in item]: pocketNames.append(('pocket' + i, int(i)))
if suff == 'pdb':
c[i] = view.add_component(filename=nglview.FileStructure(pock), **{'name': 'pocket' + i})
c[i].clear()
else:
p[i] = view.add_component(filename=nglview.FileStructure(pock), **{'name': 'pocket' + i})
p[i].clear()
# sort pocket names
pocketNames.sort(key=lambda tup: tup[1])
# representation for cavities
for pock in path_pockets_pdb:
g = re.findall('(?:pocket)(\d+)(?:_\w+)\.(\w+)', pock)
i = g[0][0]
c[i].add_surface(color='#cc0000',
radius='1.5',
lowResolution= True,
# 0: low resolution
smooth=1,
#useWorker= True,
wrap= True
)
# representation for pockets
for pock in path_pockets_pqr:
g = re.findall('(?:pocket)(\d+)(?:_\w+)\.(\w+)', pock)
i = g[0][0]
p[i].add_surface( component=i, color='skyblue', surfaceType= 'av', contour=True )
view.center()
view._remote_call('setSize', target='Widget', args=['','600px'])
view
# show pocket labels
code = """
var stage = this.stage;
var view = this.stage.viewer;
var clist_len = stage.compList.length;
var i = 0;
for(i = 0; i <= clist_len; i++){
if(stage.compList[i] != undefined && stage.compList[i].structure != undefined && stage.compList[i].parameters.ext === 'pqr') {
var elm = document.createElement("div");
elm.innerText = 'pocket' + stage.compList[i].object.name.match(/\d+/g)
elm.style.color = "black";
elm.style.background = "rgba(201, 149, 6, .8)";
elm.style.padding = "8px";
stage.compList[i].addAnnotation(stage.compList[i].structure.center, elm)
}
}
"""
view._execute_js_code(code)
view
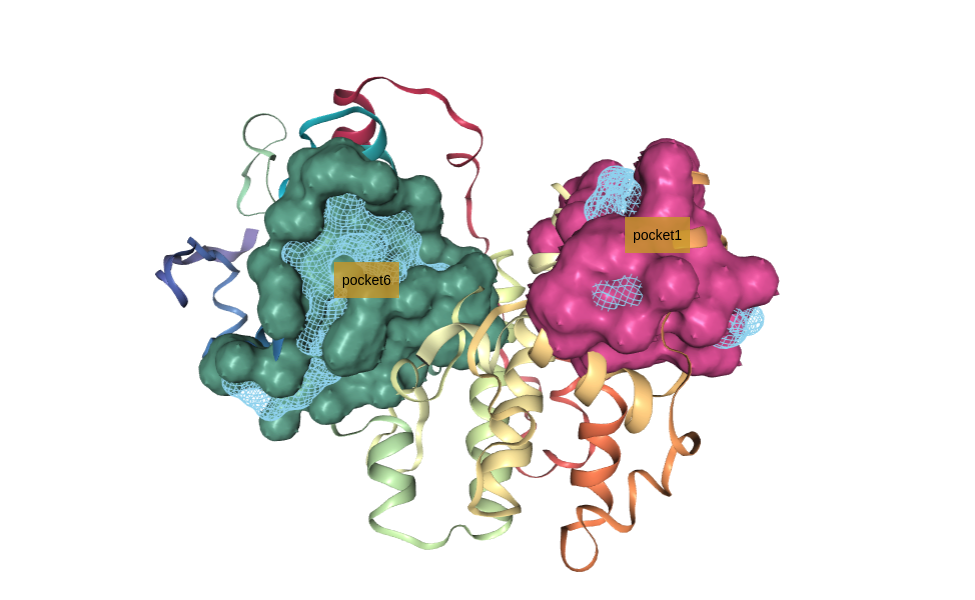
Select pocket (cavity)
Select a specific pocket (cavity) from the filtered list to be used in the docking procedure.
If fpocket has been able to identify the correct binding site, which we know from the original protein-ligand structure, it just needs to be selected. In this particular example, the pocket we are interested in is the pocket number 6.
Choose a pocket from the DropDown list:
mdsel = ipywidgets.Dropdown(
options=pocketNames,
description='Sel. pocket:',
disabled=False,
)
display(mdsel)
Extract Pocket Cavity
Extract the selected protein cavity (pocket) from the fpocket results.
It will be used to generate the docking box in the following step.
Building Blocks used:
fpocket_select from biobb_vs.fpocket.fpocket_select
from biobb_vs.fpocket.fpocket_select import fpocket_select
fpocket_cavity = "fpocket_cavity.pdb"
fpocket_pocket = "fpocket_pocket.pqr"
prop = {
"pocket": mdsel.value
}
fpocket_select(input_pockets_zip=fpocket_filter_pockets,
output_pocket_pdb = fpocket_cavity,
output_pocket_pqr=fpocket_pocket,
properties=prop)
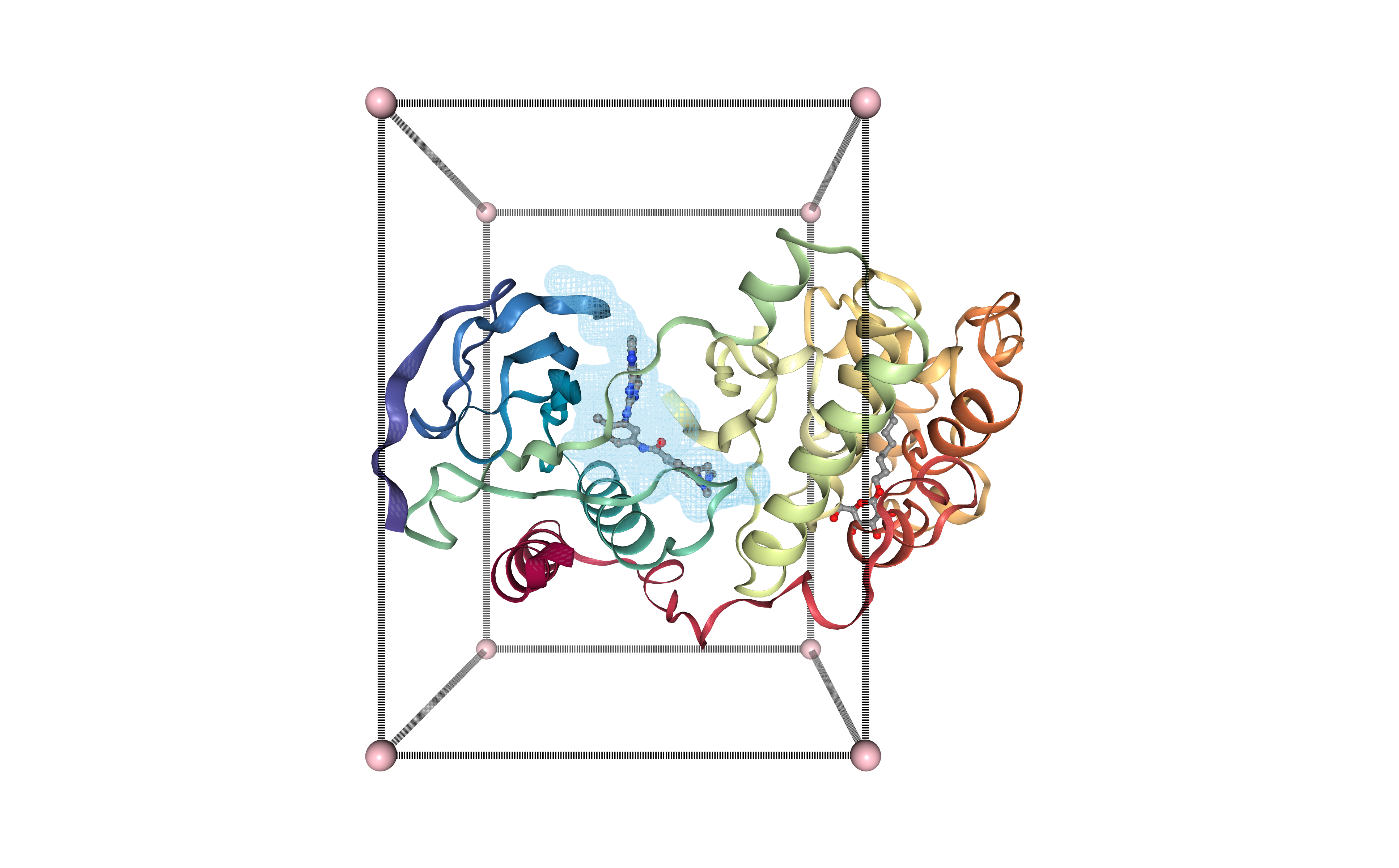
Generating Cavity Box
Generating a box surrounding the selected protein cavity (pocket), to be used in the docking procedure. The box is defining the region on the surface of the protein target where the docking program should explore a possible ligand dock.
An offset of 12 Angstroms is used to generate a big enough box to fit the small molecule and its possible rotations.
Building Blocks used:
box from biobb_vs.utils.box
from biobb_vs.utils.box import box
output_box = "box.pdb"
prop = {
"offset": 12,
"box_coordinates": True
}
box(input_pdb_path = fpocket_pocket,
output_pdb_path = output_box,
properties=prop)
Visualizing binding site box in 3D structure
Visualizing the protein structure, the selected cavity, and the generated box, all together using NGL viewer. Using the original structure with the small ligand inside (Imatinib, STI, DrugBank Ligand Code DB00619), to check that the selected cavity is placed in the same region as the original ligand.
view = nglview.NGLWidget()
s = view.add_component(nglview.FileStructure(download_pdb))
b = view.add_component(nglview.FileStructure(output_box))
p = view.add_component(nglview.FileStructure(fpocket_pocket))
p.clear()
atomPair = [
[ "9999:Z.ZN1", "9999:Z.ZN2" ],
[ "9999:Z.ZN2", "9999:Z.ZN4" ],
[ "9999:Z.ZN4", "9999:Z.ZN3" ],
[ "9999:Z.ZN3", "9999:Z.ZN1" ],
[ "9999:Z.ZN5", "9999:Z.ZN6" ],
[ "9999:Z.ZN6", "9999:Z.ZN8" ],
[ "9999:Z.ZN8", "9999:Z.ZN7" ],
[ "9999:Z.ZN7", "9999:Z.ZN5" ],
[ "9999:Z.ZN1", "9999:Z.ZN5" ],
[ "9999:Z.ZN2", "9999:Z.ZN6" ],
[ "9999:Z.ZN3", "9999:Z.ZN7" ],
[ "9999:Z.ZN4", "9999:Z.ZN8" ]
]
# structure
s.add_representation(repr_type='cartoon',
selection='not het',
color='#cccccc',
opacity=.2)
# ligands box
b.add_representation(repr_type='ball+stick',
selection='9999',
color='pink',
aspectRatio = 8)
# lines box
b.add_representation(repr_type='distance',
atomPair= atomPair,
labelVisible=False,
color= 'black')
# pocket
p.add_surface(component=mdsel.value,
color='skyblue',
surfaceType= 'av',
lowResolution= True,
# 0: low resolution
smooth=1,
contour=True,
opacity=0.4,
#useWorker= True,
wrap= True )
view.center()
view._remote_call('setSize', target='Widget', args=['','600px'])
view
Downloading Small Molecule
Downloading the desired small molecule to be used in the docking procedure.
In this particular example, the small molecule of interest is the FDA-approved drug Imatinib, with PDB code STI.
Building Blocks used:
ideal_sdf from biobb_io.api.ideal_sdf
from biobb_io.api.ideal_sdf import ideal_sdf
sdf_ideal = "ideal.sdf"
prop = {
"ligand_code": ligand_code
}
ideal_sdf(output_sdf_path=sdf_ideal,
properties=prop)
Converting Small Molecule
Converting the desired small molecule to be used in the docking procedure, from SDF format to PDB format using the OpenBabel chemoinformatics tool.
Building Blocks used:
babel_convert from biobb_chemistry.babelm.babel_convert
from biobb_chemistry.babelm.babel_convert import babel_convert
ligand = "ligand.pdb"
prop = {
"input_format": "sdf",
"output_format": "pdb",
"binary_path": "obabel"
}
babel_convert(input_path = sdf_ideal,
output_path = ligand,
properties=prop)
Preparing Small Molecule (ligand) for Docking
Preparing the small molecule structure for the docking procedure. Converting the PDB file to a PDBQT file format (AutoDock PDBQT: Protein Data Bank, with Partial Charges (Q), & Atom Types (T)), needed by AutoDock Vina.
The process adds partial charges and atom types to every atom. Besides, the ligand flexibility is also defined in the information contained in the file. The concept of “torsion tree” is used to represent the rigid and rotatable pieces of the ligand. A rigid piece (”root”) is defined, with zero or more rotatable pieces (”branches”), hanging from the root, and defining the rotatable bonds.
More info about PDBQT file format can be found in the AutoDock FAQ pages.
Building Blocks used:
babel_convert from biobb_chemistry.babelm.babel_convert
from biobb_chemistry.babelm.babel_convert import babel_convert
prep_ligand = "prep_ligand.pdbqt"
prop = {
"input_format": "pdb",
"output_format": "pdbqt",
"binary_path": "obabel"
}
babel_convert(input_path = ligand,
output_path = prep_ligand,
properties=prop)
Visualizing small molecule (drug)
Visualizing the desired drug to be docked to the target protein, using NGL viewer.
Left panel: PDB-formatted file, with all hydrogen atoms.
Right panel: PDBqt-formatted file (AutoDock Vina-compatible), with united atom model (only polar hydrogens are placed in the structures to correctly type heavy atoms as hydrogen bond donors).
from ipywidgets import HBox
v0 = nglview.show_structure_file(ligand)
v1 = nglview.show_structure_file(prep_ligand)
v0._set_size('500px', '')
v1._set_size('500px', '')
def on_change(change):
v1._set_camera_orientation(change['new'])
v0.observe(on_change, ['_camera_orientation'])
HBox([v0, v1])

Preparing Target Protein for Docking
Preparing the target protein structure for the docking procedure. Converting the PDB file to a PDBqt file, needed by AutoDock Vina. Similarly to the previous step, the process adds partial charges and atom types to every target protein atom. In this case, however, we are not taking into account receptor flexibility, although Autodock Vina allows some limited flexibility of selected receptor side chains (see the documentation).
Building Blocks used:
str_check_add_hydrogens from biobb_structure_utils.utils.str_check_add_hydrogens
from biobb_structure_utils.utils.str_check_add_hydrogens import str_check_add_hydrogens
prep_receptor = "prep_receptor.pdbqt"
prop = {
"charges": True,
"mode": "auto"
}
str_check_add_hydrogens(input_structure_path = pdb_protein,
output_structure_path = prep_receptor,
properties=prop)
Running the Docking
Running the docking process with the prepared files:
ligand
target protein
binding site box
using AutoDock Vina.
Building Blocks used:
autodock_vina_run from biobb_vs.vina.autodock_vina_run
from biobb_vs.vina.autodock_vina_run import autodock_vina_run
output_vina_pdbqt = "output_vina.pdbqt"
output_vina_log = "output_vina.log"
prop = { }
autodock_vina_run(input_ligand_pdbqt_path = prep_ligand,
input_receptor_pdbqt_path = prep_receptor,
input_box_path = output_box,
output_pdbqt_path = output_vina_pdbqt,
output_log_path = output_vina_log,
properties = prop)
Visualizing docking output poses
Visualizing the generated docking poses for the ligand, using NGL viewer.
Left panel: Docking poses displayed with atoms coloured by partial charges and licorice representation.
Right panel: Docking poses displayed with atoms coloured by element and ball-and-stick representation.
from ipywidgets import HBox
models = 'all'
v0 = nglview.show_structure_file(output_vina_pdbqt, default=False)
v0.add_representation(repr_type='licorice',
selection=models,
colorScheme= 'partialCharge')
v0.center()
v1 = nglview.show_structure_file(output_vina_pdbqt, default=False)
v1.add_representation(repr_type='ball+stick',
selection=models)
v1.center()
v0._set_size('500px', '')
v1._set_size('500px', '')
def on_change(change):
v1._set_camera_orientation(change['new'])
v0.observe(on_change, ['_camera_orientation'])
HBox([v0, v1])

Select Docking Pose
Select a specific docking pose from the output list for visual inspection.
Choose a docking pose from the DropDown list.
from Bio.PDB import PDBParser
parser = PDBParser(QUIET = True)
structure = parser.get_structure("protein", output_vina_pdbqt)
models = []
for i, m in enumerate(structure):
models.append(('model' + str(i), i))
mdsel = ipywidgets.Dropdown(
options=models,
description='Sel. model:',
disabled=False,
)
display(mdsel)
Extract a Docking Pose
Extract a specific docking pose from the docking outputs.
Building Blocks used:
extract_model_pdbqt from biobb_vs.utils.extract_model_pdbqt
from biobb_vs.utils.extract_model_pdbqt import extract_model_pdbqt
output_pdbqt_model = "output_model.pdbqt"
prop = {
"model": mdsel.value + 1
}
extract_model_pdbqt(input_pdbqt_path = output_vina_pdbqt,
output_pdbqt_path = output_pdbqt_model,
properties=prop)
Converting Ligand Pose to PDB format
Converting ligand pose to PDB format.
Building Blocks used:
babel_convert from biobb_chemistry.babelm.babel_convert
from biobb_chemistry.babelm.babel_convert import babel_convert
output_pdb_model = "output_model.pdb"
prop = {
"input_format": "pdbqt",
"output_format": "pdb",
"binary_path": "obabel"
}
babel_convert(input_path = output_pdbqt_model,
output_path = output_pdb_model,
properties=prop)
Superposing Ligand Pose to the Target Protein Structure
Superposing ligand pose to the target protein structure, in order to see the protein-ligand docking conformation.
Building a new PDB file with both target and ligand (binding pose) structures.
Building Blocks used:
cat_pdb from biobb_structure_utils.utils.cat_pdb
from biobb_structure_utils.utils.cat_pdb import cat_pdb
output_structure = "output_structure.pdb"
cat_pdb(input_structure1 = pdb_protein,
input_structure2 = output_pdb_model,
output_structure_path = output_structure)
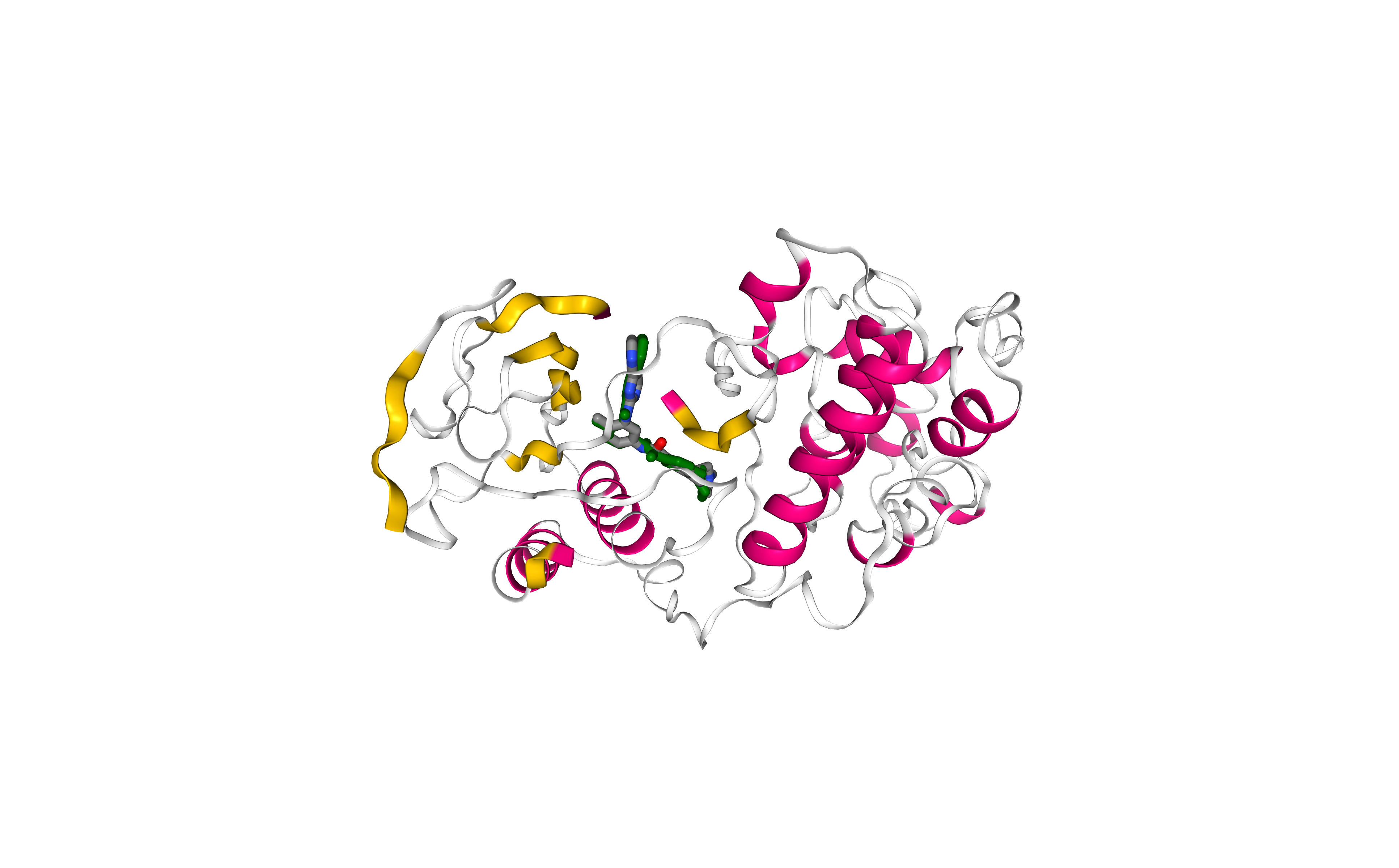
Comparing final result with experimental structure
Visualizing and comparing the generated protein-ligand complex with the original protein-ligand conformation (downloaded from the PDB database), using NGL viewer.
Licorice, element-colored representation: Experimental pose.
Licorice, green-colored representation: Docking pose.
Note that outputs from AutoDock Vina don’t contain all the atoms, as the program works with a united-atom representation (i.e. only polar hydrogens).
view = nglview.NGLWidget()
# v1 = Experimental Structure
#v1 = view.add_component(download_pdb)
v1 = view.add_component(nglview.FileStructure(download_pdb))
v1.clear()
v1.add_representation(repr_type='licorice',
selection='STI',
radius=0.5)
# v2 = Docking result
#v2 = view.add_component(output_structure)
v2 = view.add_component(nglview.FileStructure(output_structure))
v2.clear()
v2.add_representation(repr_type='cartoon', colorScheme = 'sstruc')
v2.add_representation(repr_type='licorice', radius=0.5, color= 'green', selection='UNL')
view._remote_call('setSize', target='Widget', args=['','600px'])
view
# align reference and output
code = """
var stage = this.stage;
var clist_len = stage.compList.length;
var i = 0;
var s = [];
for(i = 0; i <= clist_len; i++){
if(stage.compList[i] != undefined && stage.compList[i].structure != undefined) {
s.push(stage.compList[i])
}
}
NGL.superpose(s[0].structure, s[1].structure, true, ".CA")
s[ 0 ].updateRepresentations({ position: true })
s[ 0 ].autoView()
"""
view._execute_js_code(code)
view
Questions & Comments
Questions, issues, suggestions and comments are really welcome!
GitHub issues:
BioExcel forum: